How to perform Nucleus Independent Chemical Shift Calculations using CASTEP and Pynics
Written by Ben Tatman
Last revised: 09/05/2022
The chemical shift at which a nuclear environment appears in solid-state NMR is dependent not only on the local intramolecular interactions affecting that environment, but also on interactions with longer range features, such as ring currents and hydrogen bonding. Using DFT GIPAW calculations, it is possible to partition the contributions of these different features to chemical shift. Further, it is possible to observe the spatial effects of these contributions, and the distance at which these contributions are significant.
Zilka et al. 20171 and Sturniolo et al. 20192 have developed techniques to partition these different contributions to the chemical shift. Here, scripts to perform this analysis will be demonstrated. The scripts have been written using python 3 and are available via the pynics package. pynics can simply be installed via pip:
pip install --user pynicsThis command will automatically install numpy, ASE (Atomic Simulation Environment), matplotlib and Soprano if they're not already present. It will also place on your PATH two scripts: splitcell and nicsanalyse which will be used below. See GitHub for more information on installing pynics in a separate python environment.
Supercell Preparation
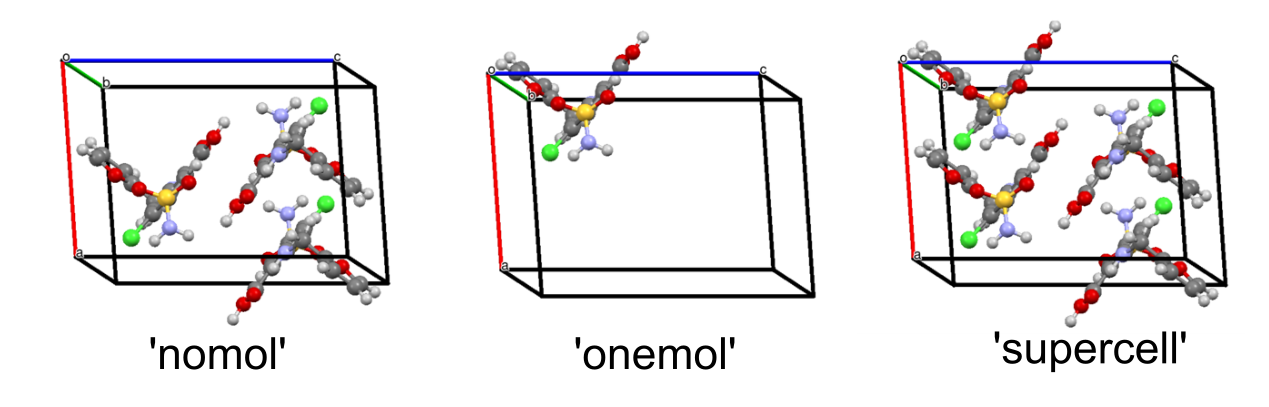
The technique introduced by Zilka et al. 20171 involves creating three different CASTEP .cell input files representing different components of the system: a supercell (supercell), an isolated molecule (onemol), and a supercell missing a molecule (nomol). These should be derived from a single supercell, as detailed here.
First, a single unit cell of the desired structure should be geometry optimized using CASTEP. The procedure to do this has been given in detail elsewhere and so shall not be repeated here.
A tool such as Mercury should be used to determine the size of supercell required. Ideally, this will have a central molecule with a separation > 10 Å from where it would repeat outside the supercell.3 If, for example, a 2 × 2 × 1 supercell is required, the cif2cell utility may be used to create a supercell;
cif2cell --program=CASTEP --no-reduce --supercell=[2,2,1] -f SEED.cif -o SEED.cellThe argument --program=CASTEP tells it to generate a cell file compatible with CASTEP, --no-reduce makes it not reduce the unit cell, and --supercell provides the supercell parameters (e.g. number of unit cells along each axis).
Onemol and Nomol Preparation
The splitcell utility may be used to split the supercell into a 'one mol', 'no mol', and 'supercell' file (see examples for furosemide in Figure 1). This may be invoked as
splitcell --struct <structure file> --nicslist <file to output nicslist>This will extract a molecule from the provided structure file (SEED.cell) and save this in SEED_onemol.cell, output the remaining structure in SEED_nomol.cell, and the supercell in SEED_supercell.cell. Note that the supercell output by this script must be used as opposed to the initial SEED.cell file, as there may be subtle differences between them arising from the processing by ASE and Soprano. Additionally, a nicslist is output for use later. This contains the positions of the nuclei for which the nucleus independent chemical shift will be calculated.
Magres Calculations
CASTEP GIPAW calculations should then be carried out on these cell files. This should be performed using a version of CASTEP >5, with MAGRES_WRITE_RESPONSE=True added to the .param file. Note: This will produce a current.dat file regardless of the SEED name, and so it is recommended to perform each in a separate directory to ensure they don\'t overwrite one another.
Analysing NICS
To analyse the resulting magres and current.dat files, a script called nicsanalyse.py has been created and may be run as:
nicsanalyse --nicslist <path to nicslist> \\
--ref <reference shielding value> \\
--nomol_magres <path to nomol magres> \\
--onemol_magres <path to onemol magres> \\
--supercell_magres <path to supercell magres> \\
--output <output file>where it is assumed that the magres files and associated current.dat files are within the same directories. Additional options exist to produce build up curves for the magnetic shielding as a function of distance from the nucleus of interest (as found in Sturniolo et al. (2019)2). These may be found by passing the -h parameter, or by passing no parameters.
This will output a text file containing a table with the different NICS parameters; the chemical shifts for both the supercell and isolated molecule, the NICS shift, and the electronic structure shielding contribution. An example for the furosemide structure (Figure 1) is shown in the table below, with the data provided here. This table may be compared to Table 6 by Zilka et al. (2019),4 with small differences likely arising from different choices of energy cutoffs in the GIPAW calculations.
| # Atom | Type | # in prior work4 | δsupercell / ppm | δonemol / ppm | NICS / ppm | Δδ / ppm | δes / ppm |
|---|---|---|---|---|---|---|---|
| 1 | CH | 11 | 6.86 | 6.44 | -0.28 | 0.41 | 0.70 |
| 2 | CH | 10 | 5.73 | 5.29 | 0.00 | 0.43 | 0.44 |
| 3 | CH | 2 | 5.49 | 5.25 | -0.19 | 0.23 | 0.43 |
| 4 | CH2 | 7 | 2.03 | 3.38 | 1.47 | -1.34 | -2.81 |
| 5 | CH2 | 8 | 4.12 | 4.65 | 0.36 | -0.53 | -0.89 |
| 6 | NH | 2 | 7.91 | 8.37 | 0.11 | -0.46 | -0.57 |
| 7 | CH | 3 | 5.27 | 5.85 | 0.56 | -0.59 | -1.15 |
| 8 | CH | 6 | 7.49 | 7.48 | -0.04 | 0.01 | 0.05 |
| 9 | OH | 1 | 13.14 | 5.55 | 1.52 | 7.59 | 6.07 |
| 10 | NH2 | 4 | 5.91 | 2.98 | -0.34 | 2.93 | 3.27 |
| 11 | NH2 | 5 | 4.94 | 4.31 | -0.03 | 0.63 | 0.67 |
nicsanalyse for the furosemide (CCDC:FURSEM16) system (see Figure 1). The chemical shifts, δsupercell and δonemol were calculated using Equation 1 with a reference shielding of 29.30 ppm.The chemical shift of a site in a crystalline solid arises both due to intramolecular and intermolecular effects. The chemical shift may be calculated from the GIPAW calculated magnetic shielding as
\(\begin{equation} \delta = \sigma_{\text{ref}} - \sigma, \tag{1}\\ \end{equation}\)
where \(\sigma\)ref is the reference shielding (here taken as 29.30 ppm). The chemical shift may be calculated both for a supercell, \(\delta\)supercell, and a single isolated molecule, \(\delta\)onemol. The molecule to crystal change in chemical shift, \(\Delta \delta\), may be calculated as
\(\begin{equation} \Delta\delta = \delta_{\mathrm{supercell}} - \delta_{\mathrm{onemol}}.\tag{2} \\ \end{equation}\)
The change in chemical shift from a molecule to crystal originates from intermolecular interactions, which may be thought to arise due to two different effects. Firstly, intermolecular ring currents may lead to a change in the current density at a point in the molecule, leading to a nucleus independent chemical shift, NICS. We may calculate this using the current response from the supercell system in which the molecule of interest has been removed,1,2 as was performed using the 'nomol' magres calculation. Secondly, the presence of intermolecular interactions such as hydrogen bonding may lead to changes in the local electronic structure of the molecule. This 'electronic structure' contribution may be calculated as
\(\begin{equation} \delta_{\mathrm{ES}} = \delta_{\mathrm{supercell}} - \delta_{\mathrm{\mathrm{onemol}}} - \mathrm{NICS}\ = \Delta\delta - \mathrm{NICS}\tag{3}, \end{equation}\)
and can provide information as to the influence of hydrogen bonding. The resulting output of the nicsanalyse script will produce a table like that in Table 1 containing these contributions, which may provide insight into the hydrogen bonding or other intermolecular bonding interactions occurring within the crystal. Further detail regarding these calculations and for interpretation of the output of the scripts used here may be found in Refs 1, 2, and 4.
- Zilka, M., Sturniolo, S., Brown, S. P. & Yates, J. R. Visualising crystal packing interactions in solid-state NMR: Concepts and applications. J. Chem. Phys. 147, 144203 (2017).
- Sturniolo, S. & Yates, J. R. The Lorentz sphere visualised. J. Chem. Phys. 150, 094103 (2019).
- Tatton, A. S. et al. Isolated molecule calculation protocol with Materials Studio.
- Zilka, M., Yates, J. R. & Brown, S. P. An NMR crystallography investigation of furosemide. Magn. Reson. Chem. 57, 191-199 (2019).